


L’Istituto Zooprofilattico Sperimentale della Venezie (IZSVe) ha sequenziato l’intero genoma del virus SARS-CoV-2 responsabile di casi di COVID-19 in Veneto. Il sequenziamento è stato eseguito a partire da 13 tamponi naso/faringei inviati dall’Az. ULSS 9 di Verona, con cui l’IZSVe ha stretto un accordo di collaborazione scientifica. Le sequenze sono state depositate nel database pubblico GISAID, rendendo così disponibili per la comunità scientifica le prime sequenze di SARS-CoV-2 dal Veneto.
SARS-CoV-2 appartiene al genere Betacoronavirus, di cui fanno parte anche i virus SARS-CoV e MERS-CoV. Con un genoma di oltre 29.000 basi è uno dei virus a RNA con il genoma più lungo e complesso (fig. 1), in grado di evolvere sia attraverso la comparsa di mutazioni che mediante eventi di ricombinazione genetica.
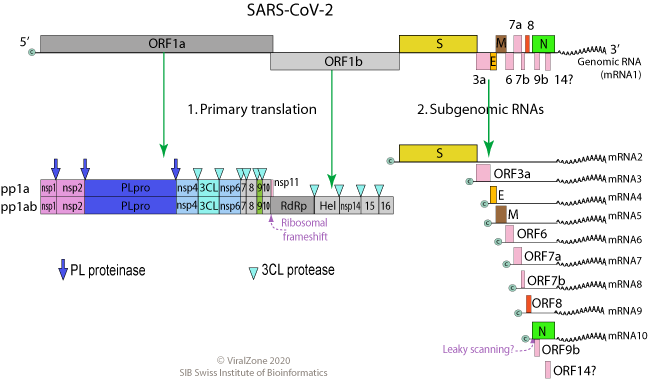
Figura 1. Organizzazione del genoma del virus SARS-CoV-2 (ViralZone, Swiss Institute of Bioinformatic, https://viralzone.expasy.org/9076)
Le sequenze genomiche di SARS-CoV-2 sono state classificate in due gruppi (chiamati lineage) principali: A e B. Il lineage A si distingue in 5 ulteriori lineage (A.1-A.5) e due sub-lineage, mentre nel lineage B si distinguono 9 lineage (B.1-B.9) e molteplici sub-lineage (Fig. 2).
I virus italiani (in blu e rosso nell’albero filogenetico) cadono all’interno dei lineage B, B.1 e B.1.5. I virus B derivano dai campioni prelevati dalla coppia di turisti cinese ricoverata a gennaio a Roma, mentre i virus circolanti nella popolazione italiana da metà febbraio appartengono al lineage B1 o al sub-lineage B.1.5. Tutti i virus di Verona (in rosso in Fig. 2) appartengono al lineage B1.
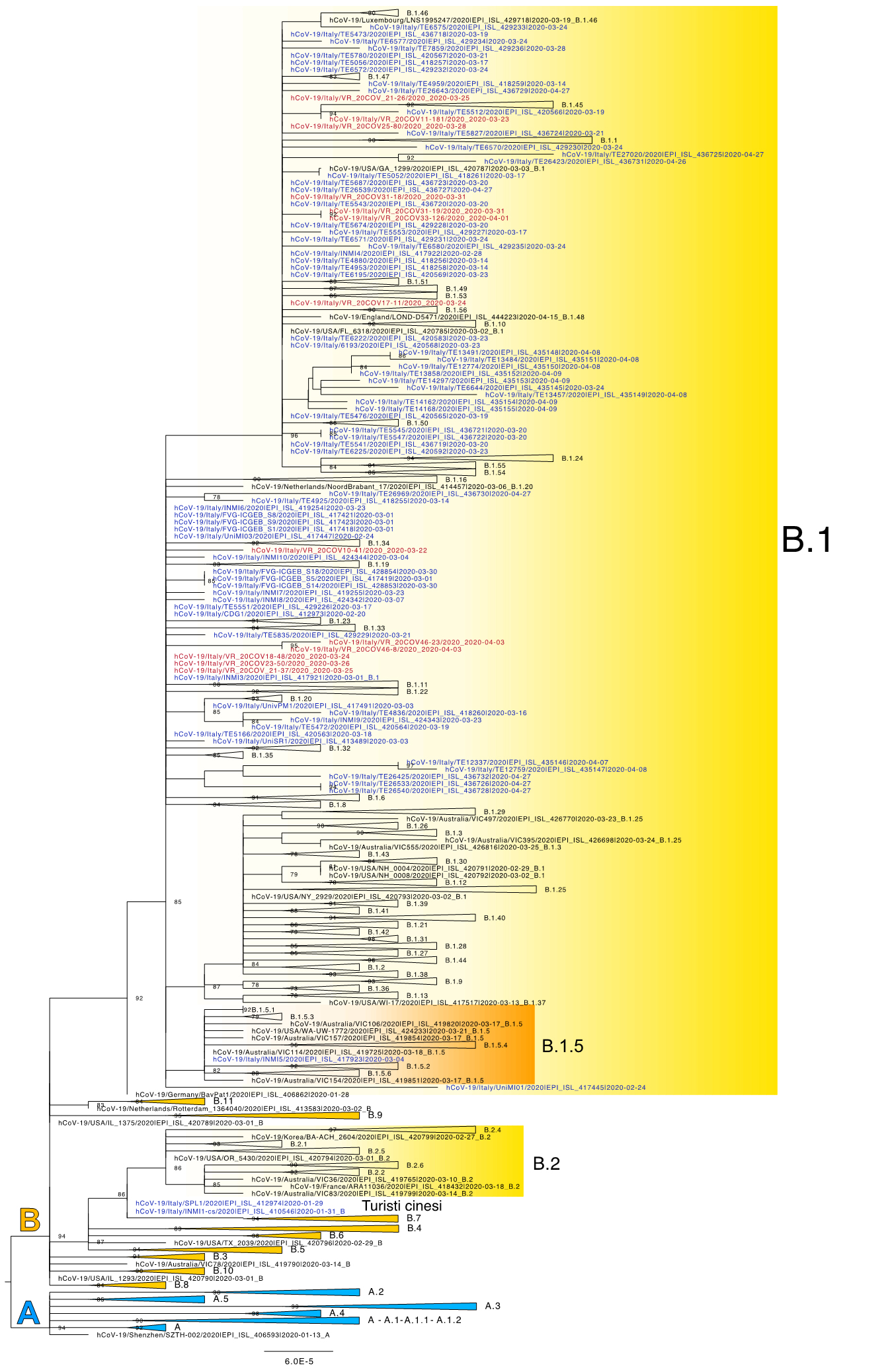
Figura 2. Albero filogenetico del virus SARS-CoV-2. Le sequenze del Veneto (provincia di Verona) sono in rosso, mentre le sequenze dalle altre regioni italiane sono in blu. I diversi lineage e sub-lineage identificati ad oggi sono evidenziati nell’albero. I virus italiani appartengono al lineage B, B.1, B.1.5.
Il network del genoma (Figure 3A e 3B) è stato ottenuto dalle sequenze del genoma di tutti i virus italiani disponili ad oggi. Tale analisi mostra la circolazione in Italia di molteplici varianti virali, strettamente correlate tra di loro, che si distinguono per 1-17 differenze nucleotidiche. Tra queste, due mostrano una frequenza maggiore nella popolazione e sembrano aver circolato estensivamente in diverse regioni italiane (Fig. 3A) sin dall’inizio dell’epidemia. Entrambe le varianti sono state identificate in Veneto (provincia di Verona). È interessante notare che la maggior parte delle sequenze più recenti (aprile) si discosta da queste due varianti per una o più sostituzioni nucleotidiche (Fig. 3B).
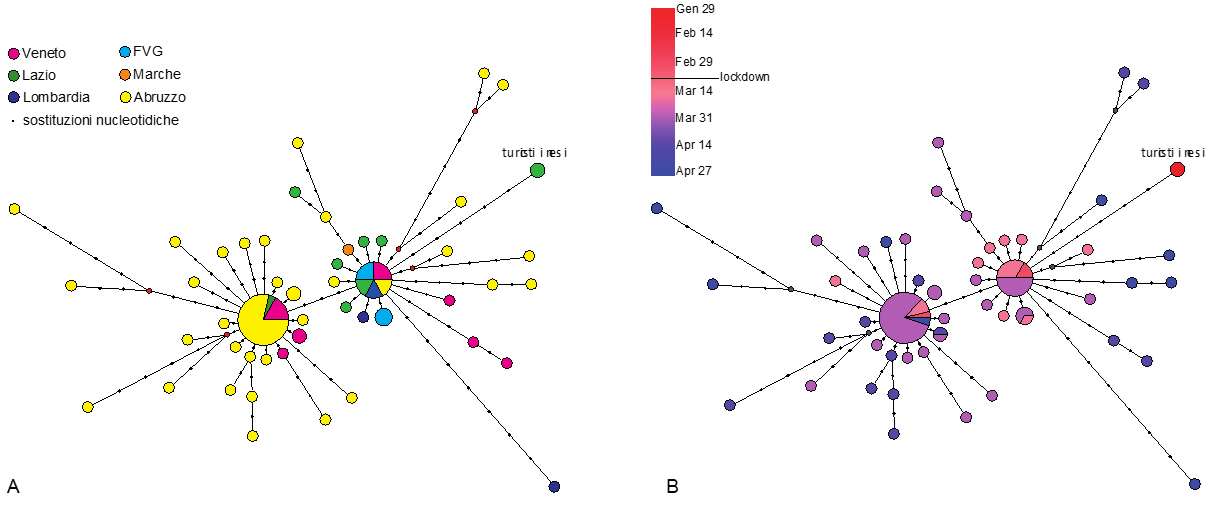
Figura 3. Network del genoma delle varianti di SARS-CoV-2 identificate in Italia. Ciascun cerchio rappresenta un genotipo (cioè una particolare variante del virus). Le dimensioni del cerchio sono proporzionali al numero di virus sequenziati appartenenti a quella variante. Le braccia che connettono i cerchi hanno una lunghezza proporzionale al numero di differenze nucleotidiche che distinguono una variante dall’altra. I colori corrispondono alle regioni di campionamento del virus (A) o alla data di campionamento (B).
Tutti i campioni italiani B1 presentano una mutazione (D614G) a livello della proteina spike, la proteina che permette al coronavirus di infettare le cellule umane. Questa mutazione è presente anche in molti dei virus circolati in Europa, dove è diventata molto velocemente la forma virale predominante. Secondo uno studio recente (non sottoposto a peer review) potrebbe facilitare l’entrata del virus nelle cellule dell’ospite (Bhattacharyya et al., 2020. Anteprima disponibile in: https://doi.org/10.1101/2020.05.04.075911).
Le piccole differenze osservabili nei diversi ceppi del SARS-CoV 2 sono tipiche dei virus ad RNA, che cambiano molto facilmente nel passaggio da un soggetto all’altro, e sono utili per tracciare dinamiche geografiche e comprendere eventuali connessioni epidemiologiche.
Sequenziare regolarmente il genoma di questi virus è fondamentale per monitorarne l’evoluzione, indagare l’acquisizione di mutazioni che potrebbero determinare cambiamenti delle proprietà antigeniche (e quindi ridurre la protezione immunitaria precedentemente acquisita) o della virulenza. Attualmente non ci sono elementi che ci consentono di dire che i SARS-CoV-2 circolanti abbiano cambiato il loro potere patogeno, ossia che siano diventati più o meno aggressivi.
Ad oggi ci sono più di 19.000 sequenze dell’intero genoma di SARS-CoV-2 disponibili in database pubblici, ma solo 96 di queste sequenze sono italiane. L’IZSVe è pronto a mettere a disposizione le proprie competenze per la caratterizzazione del genoma di altri SARS-CoV-2 dalla nostra Regione.



{kind=link}
{kind=link}
{kind=link}
{kind=link}