


L’Istituto Zooprofilattico Sperimentale della Venezie (IZSVe) ha sequenziato il genoma completo di quattro campioni di SARS-CoV-2 ricevuti il 3 e 4 luglio 2020, provenienti dalle province di Padova e Vicenza. Le sequenze dei virus risultano identiche tra loro e appartengono al cluster dei virus identificati in Serbia, ben diversi dai virus identificati finora in Veneto e in Italia, a indicare una probabile connessione epidemiologica tra i quattro casi.

L’Istituto Zooprofilattico Sperimentale della Venezie (IZSVe) ha sequenziato il genoma completo di quattro campioni di SARS-CoV-2 ricevuti il 3 e 4 luglio 2020, provenienti dalle province di Padova e Vicenza. Le sequenze dei virus risultano identiche tra loro e appartengono al cluster dei virus identificati in Serbia. Dalle analisi effettuate per identificare il virus nei campioni clinici è emerso che la carica virale era molto elevata, tuttavia alle specifiche mutazioni riscontrate non è possibile associare una diversa (maggiore o minore) patogenicità del virus.
Dalle analisi effettuate per identificare il virus nei campioni clinici è emerso che la carica virale era molto elevata, tuttavia alle specifiche mutazioni riscontrate non è possibile associare una diversa (maggiore o minore) patogenicità del virus, poiché finora non ci sono sufficienti studi che mettano in relazione le forme cliniche con le mutazioni osservate.
I quattro virus sequenziati si differenziano dalle varianti identificate ad oggi in Veneto e in Italia, poiché mostrano da 7 a 72 differenze nucleotidiche con i virus del Veneto e da 7 a 165 differenze nucleotidiche con i virus identificati in altre regioni d’Italia.
Sulla base dei dati genetici ad oggi disponibili, la stretta correlazione dei virus di Padova e Vicenza di luglio con i virus della Serbia suggerisce che la nuova variante possa essere stata introdotta in Italia da persone che hanno soggiornato in questo paese. L’incrocio di queste analisi con dati epidemiologici potrà supportare tale ipotesi.
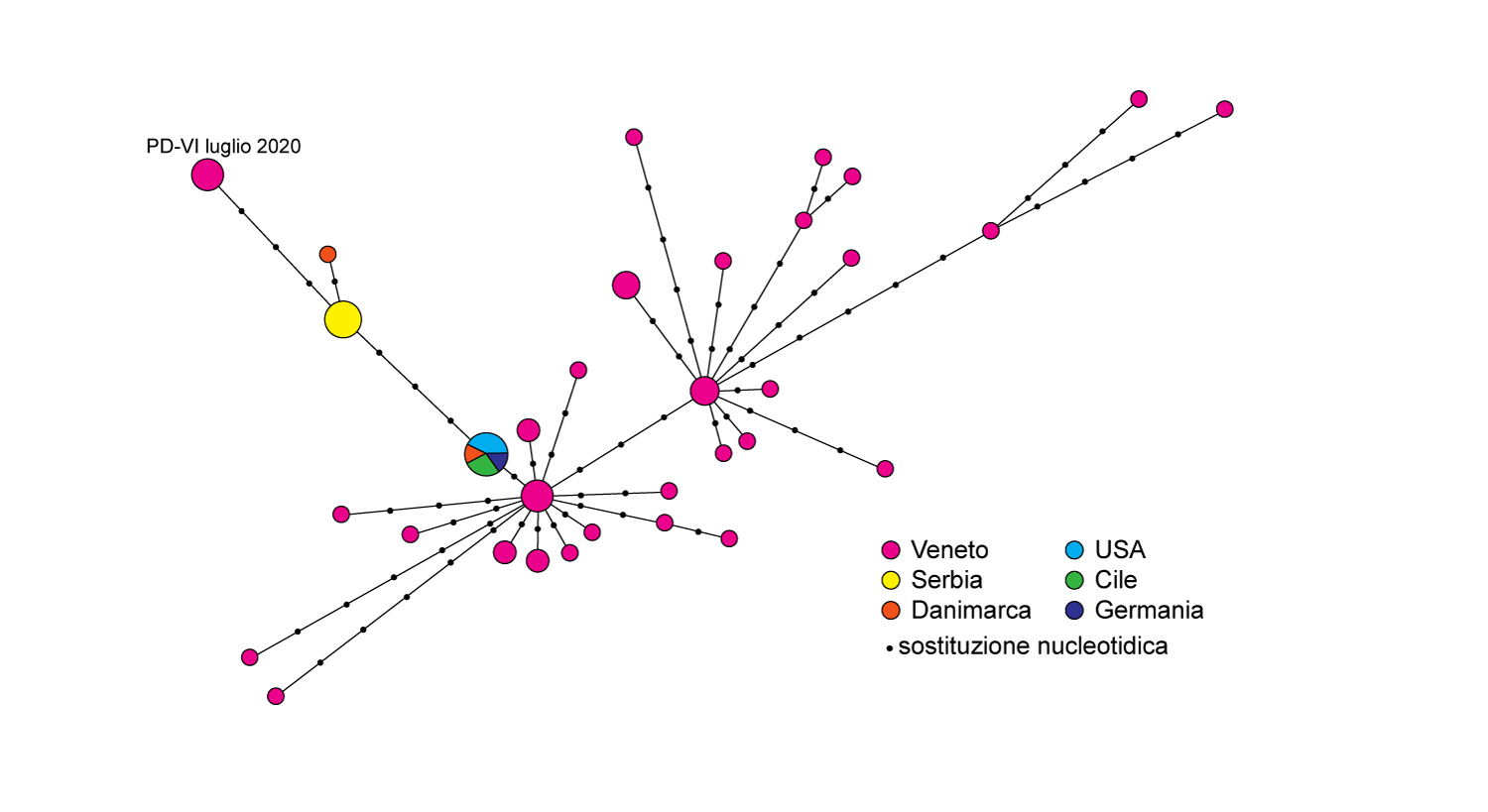
L’analisi filogenetica (Figura 1) mostra che i virus analizzati appartengono al lineage B.1.1. Le sequenze sono strettamente correlate con quelle di virus identificati in Serbia e in Danimarca nei mesi scorsi. Tale correlazione è confermata dal network riportato in Figura 2. In particolare, la variante genetica identificata nei campioni di Padova e Vicenza mostra 3 differenze nucleotidiche rispetto ai virus dalla Serbia e 4 differenze nucleotidiche rispetto al virus dalla Danimarca.
Il lineage B.1.1 è stato identificato anche in Italia e in Veneto. Infatti, le sequenze di virus Italiani (in blu in Figura 1) ricadono all’interno di quattro lineage nominati B, B.1, B.1.1 e B.1.5. Le 39 sequenze ottenute ad oggi in Veneto (in fucsia in Figura 1) da tamponi effettuati nel periodo 21 marzo-1 maggio si distribuiscono in tutti e quattro i gruppi. Tali gruppi genetici sono stati identificati anche in altri paesi Europei e in varie parti del mondo (in nero in Figura 1).
Figure (cliccare per ingrandirle)
Figura 1. Albero filogenetico del genoma completo del virus SARS-CoV-2 (metodo della massima verosimiglianza). Le sequenze italiane sono identificate in blu, le sequenze del Veneto sono in fucsia. I diversi lineage e sub-lineage identificati ad oggi in Italia sono evidenziati nell’albero. Il riquadro azzurro identifica il cluster genetico dei virus di Padova/Vicenza (luglio 2020), Serbia (aprile 2020) e Danimarca (marzo 2020).

Figura 2. Network del genoma delle varianti di SARS-CoV-2 identificate in Veneto e delle sequenze che si raggruppano con i virus identificati a Padova e Vicenza nel mese di luglio 2020. Ciascun cerchio rappresenta un genotipo (cioè una particolare variante del virus) e le dimensioni del cerchio sono proporzionali al numero di virus sequenziati appartenenti a quella variante. I colori corrispondono alle zone di campionamento del virus. Le braccia, che connettono i cerchi, hanno una lunghezza proporzionale al numero di differenze nucleotidiche che distinguono una variante dall’altra.
Si ringraziano tutti gli autori che hanno depositato e condiviso i dati genetici in GISAID (https://platform.gisaid.org/). Le analisi si basano sui dati disponibili nel database in data 09/07/2020.



{kind=link}
{kind=link}
{kind=link}
{kind=link}