


L’ondata epidemica di influenza aviaria del 2016-2017 è stata la più vasta degli ultimi dieci anni in Europa. Questa volta a colpire è stato il ceppo H5N8 ad alta patogenicità (HPAI), che ha causato più di 1.200 focolai negli allevamenti domestici, con mortalità e danni economici elevatissimi, e coinvolto migliaia di volatili selvatici.
L’Italia è stata fortemente coinvolta con 2,7 milioni di volatili interessati in 83 focolai di infezione, concentrati nelle regioni a più alta densità di popolazione avicola, Lombardia, Emilia Romagna e Veneto. Rispetto al resto d’Europa, la situazione italiana ha presentato una caratteristica singolare. Infatti la dinamica di diffusione del virus è avvenuta in due ondate: la prima tra dicembre 2016 e maggio 2017, la seconda tra luglio e dicembre 2017, quest’ultima ben più violenta con 67 focolai (80% del totale).
Si è trattato di un fenomeno molto complesso dal punto di vista sanitario che ha richiesto ingenti risorse economiche: solo in Italia le spese di eradicazione si sono aggirate intorno ai 40 milioni di euro. Uno dei problemi maggiori in casi come questo è la tempestività degli interventi per il contenimento dell’epidemia e la capacità di implementare una strategia di risposta rapida ed efficace.
Per migliorare la gestione dell’emergenza un team di epidemiologi e virologi dell’IZSVe ha sviluppato un approccio epidemiologico basato sull’integrazione di dati epidemiologici e biomolecolari, che ha permesso di conoscere l’origine e le modalità di diffusione del virus e di orientare la strategia di eradicazione “in tempo reale”. Lo studio è stato pubblicato sulla rivista Scientific Reports.
Le informazioni raccolte “sul campo” come risultato delle indagini epidemiologiche (specie colpite, distribuzione geografica degli allevamenti, movimentazioni di persone e animali, caratteristiche ambientali, ecc.) sono state combinate con l’analisi dei dati genetici effettuata nei laboratori di virologia e biologia molecolare, allo scopo di monitorare l’evoluzione della situazione e individuare, giorno per giorno, le cause e le vie di diffusione dell’infezione. Solitamente il confronto fra dati epidemiologici e biomolecolari viene fatto a epidemia conclusa; qui invece l’identificazione delle modalità di diffusione del virus è avvenuta durante l’epidemia, tramite lo studio del “contact network”, consentendo alle autorità sanitarie di predisporre azioni correttive e misure di prevenzione più efficaci per contenere la diffusione dell’infezione negli allevamenti.
L’analisi filogenetica ha rivelato che la rete dei rapporti di “parentela genetica” tra i virus era confrontabile con quella degli allevamenti infetti: quello che era un sospetto epidemiologico è stato confermato dalla similarità genetica dei virus isolati dai diversi focolai. L’integrazione di evidenze epidemiologiche con i risultati di analisi genetiche e bioinformatiche ha permesso di avere un quadro più dettagliato dell’epidemia di H5N8 HPAI in Italia e di potenziare il sistema di prevenzione, identificando le aree in cui rendere più precise le misure di controllo e rafforzare la biosicurezza.
Leggi l’articolo su Scientific Reports »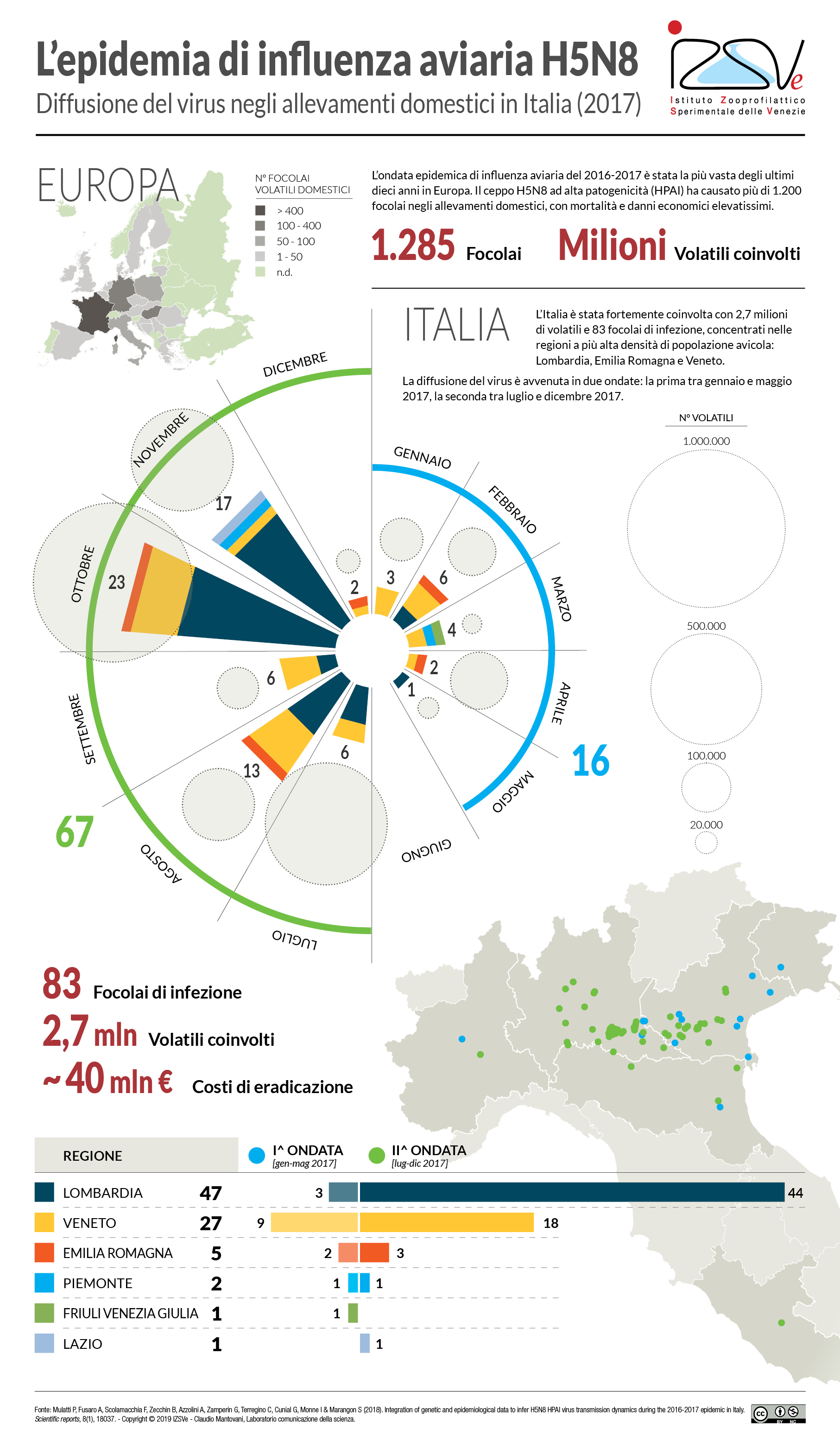



{kind=link}
{kind=link}
{kind=link}
{kind=link}